
|
|
| Home | Research | Publications | Recent news | Group | Teaching |
|
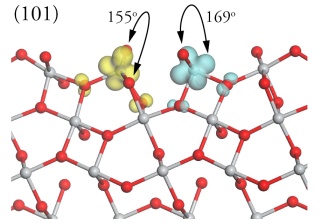
Oxide surface structure and reactivityThe structure and stability of oxide surfaces are key factors that control a wide range of technologically important processes, including sintering, catalysis and corrosion. Surface and electronic structure of TiO2The surface structure of the {110} surface of TiO2 Rutile has been studied many time using DFT. However, when the surface is reduced DFT fails to model the electronic defects and produces a delocalised electron density. The resulting metallic EDOS is incompatible with experiment and is the result of the self interaction error in DFT. DFT+U has been used to correct for this error and has allowed us to study the reduced surfaces of TiO2. As an example we show below the reduced {110} and {101} surfaces of TiO2-Rutile using DFT+U showing that DFT+U produces the correct localisation and a a defect state (in blue and yellow) which appears in the band gap. 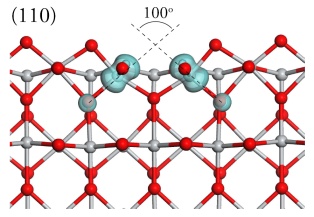
related references:
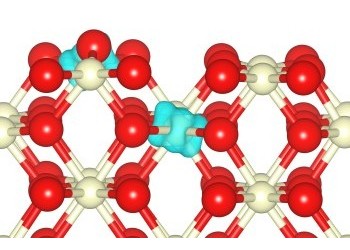
Morgan B.J. Scanlon D.O. and Watson G.W. The Use of the `+U' Correction in Describing Defect States at Metal Oxide Surfaces: Oxygen Vacancies on CeO2 and TiO2, and Li-doping of MgO. E-journal of Surface Science and Nanotechnology in press Adsorption of molecules on the low index surface of CeriaThe ability to study redox reactions allows the caclulation of the interaction of molecules with redox catalysts. We have studies the interaction of CO and NO2 with the low index surfaces of ceria and find that on adsorption a redox reaction spontaneously occurs. On CO adsorption to the stoichiometric surface the surface transfer two oxygens to the CO molcule formaing a carbonate anion while this oxidation is couple with a reduction of two surface Ce atoms from Ce(IV) to Ce(III). On NO2 adsorption on reduced Ceria surfaces an electron is transferred to the radical molecular forming NO2- while the surface is oxidized. Once again the molecular adsorption requires accurate caclualtions to obtain the geometry of the adsorbed molecule
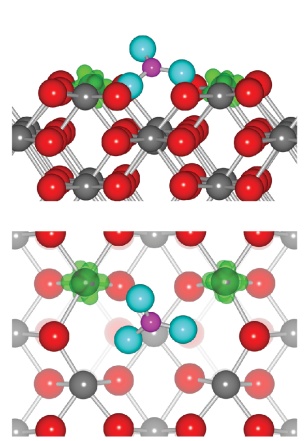
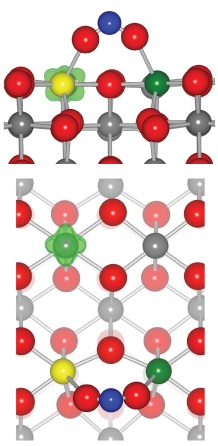
Figure showing the minimum energy adsorption modes for CO and NO2 on the (110) surface of ceria related references:
The use of DFT+U to model the reduction of the surfaces of ceria.Ceria has the unusaly property of being able to release oxygen in oxygen poor environments and taking oxygen in in oxygen rich environment. This is linked to the ability of cerium to switch oxidation state from (IV) to (III) while retaining its crystal structure (fluorite). It is vital to model the reduction process if we are to understand its role in both oxidation and redcution catalysis. Standard DFT fails to model reduced ceria due to the self interaction error (SIE). The SIE comes about due to the approximate exchange in standard DFT exchange functionals. The approximate exchange fails to cancel the electrons self Coulomb interaction and hence in DFT an electron interact with itself (this does not happen in Hartree-Fock which has exact exchange). Normally this does not present serious problems however when electronic defects are introduced these are often delocalised in DFT as the electron repels itself. This is particularly bad for localised orbitals such as d and f electrons which are high localised on their atoms. DFT when applied to reduced ceria results in a delocalised electron spread over all of the cerium atoms in the simulation cell. The EDOS shows that the electron simply occupy the bottom of the conduction band which is incosistent with experiment. FIGURE OF DFT OF REDUCED CERIA + EDOS DFT+U penalised partial electron occupations and hence encourages localisation. When DFT+U is applied to reduced cerai (U=7) the two electrons generated when the oxygen is lost are now localised on the cerium atoms adjancent to the vacancy. The EDOS now gives rise to a defect state in the vacuum gap as seen experimentally. 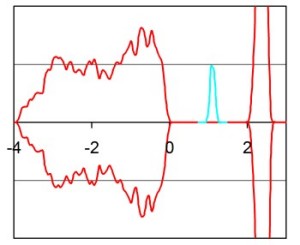
related references:
Email: watsong AT tcd.ie Last updated: Apr 19 2024 Back to Top |
