|
|
Structure and reactivity of Metal Surfaces
Metal surfaces are key components in a wide range of technologically
important materials. The reactivity is vital to processes such as sintering,
catalysis and corrosion.
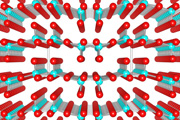
Adsorption of hydrogen to the {111} surface of Ni, Pd and Pt.
Platinum, palladium and nickel, despite their close structural relationship
and their proximity in the periodic table, show several significant differences
in their chemistry. Density functional theory calculations on the adsorption
of hydrogen onto the {111} surfaces of Ni, Pd and Pt surfaces show a considerable
difference in adsorption energies. For Ni and Pd the adsorption energies
vary as a function of the hydrogen coordination with the atop site the least
stable followed by the bridge and the two 3 fold hollow sites (hcp and fcc)
the most stable. The results are significantly different o Pt with an almost
uniform adsorption energy and no correlation to metal – hydrogen coordination.

 The similar adsorption energies for all sites on Pt imply that the diffusion
of hydrogen across the surface will be more rapid than for the other two
metals. Evidence for this effect can be found in isomerisation data. At low
partial pressure of hydrogen Pd and Ni show isomerisation of the olefin while
Pt does not. This can be explained by the rate of the hydrogenation reaction
on Pd and Ni being limited by the transport of hydrogen to the reaction site.
On Pt, due to the faster diffusion hydrogen transport will not be a significant
factor.
The similar adsorption energies for all sites on Pt imply that the diffusion
of hydrogen across the surface will be more rapid than for the other two
metals. Evidence for this effect can be found in isomerisation data. At low
partial pressure of hydrogen Pd and Ni show isomerisation of the olefin while
Pt does not. This can be explained by the rate of the hydrogenation reaction
on Pd and Ni being limited by the transport of hydrogen to the reaction site.
On Pt, due to the faster diffusion hydrogen transport will not be a significant
factor.
The results indicate that there are fundamental differences in the interaction
of hydrogen on the surfaces of platinum, paladium and nickel, and that
these differences may contribute to the differences in the hydrogenation
activity
related references:
- Watson G.W., Wells R.P.K., Willock D.J. and Hutchings G.J.
A comparison of the adsorption and diffusion of hydrogen on the {111}
surfaces of Ni, Pd, and Pt from density functional theory calculations
Journal of Physical Chemistry 105, 4889-4894 (2001)
- Watson G.W. Wells R.P.K., Willock D.J and Hutchings G.J.
Ab initio simulation of the interaction of hydrogen with the {111}
surfaces of platinum, palladium and nickel. A possible explanation for their
difference in hydrogenation activity.
Chemical Communications, 8, 705-706 (2000).
 The adsorption of ethene on the {111} surface of platinum.
The adsorption of ethene on the {111} surface of platinum.
The adsorption of hydrocarbons onto the surfaces of transition metals
is a subject of great interest to workers in both surface science and catalysis
due to its importance in many catalytic processes e.g. hydrogenation /
dehydrogenation and isomerisation. The electronic interaction of alkenes
with the surface will play a role in the bonding of reactants to the surfaces
of the catalyst. Because of this, the adsorption of ethene has been used
as a model system and has been extensively studied on a variety of transition
metals using a broad range of methods. We have employed periodic density
functional theory (DFT) with gradient corrections to study the structure
and energetics of the adsorption of ethene on the {111} surface of platinum.
Six adsorption modes shown in the figure were investigated on a rigid
3 layer periodic slab expressing the Pt {111 surface (a) cross bridge, b)
atop bridge, c) bridge, d) atop hollow, e) fcc hollowand f) hcp hollow).
The most stable site was the bridge site (di-s
type adsorption) with an adsorption energy of 108.7 kJ mol-1
and C-C bond length of to 1.483 Å, which is significantly longer
than the calculated gas phase ethene bond length of 1.334 Å. The recently
proposed fcc hollow site adsorption was found to be significantly less stable
(63.6 kJ mol -1) although slightly more favourable than the atop
( p adsorbed) modes (59 kJ mol-1).
The cross bridge site was found to be essentially unbound.
The bridge adsorption mode resulted in significant distortion of the molecule
with the carbon atoms attaining a sp3 like hybridised conformation
with the C-CH2 bond angle having reduced from 180 degrees in gas
phase ethene to 138 degrees. The pi adsorbed modes. which are important in
hydrogenation reactions, also showed significant but smaller distortion, with
a C-C bond length if 1.40 and C-CH2 angles of around 160 degrees.

The effect of surface relaxation on the adsorption energy and structure
was investigated by allowing the entire Pt {111} slab to relax giving rise
to large changes in the positions of the co-ordinating Pt atoms. The bridge
site shows displacements of 0.235 Å out of the surface for the two
Pt atoms directly co-ordinated to the ethene C atoms with an increase in
the adsorption energy of 18.6 kJ mol-1 compared to the rigid surface
case from 108.7 kJ mol-1 to 127.3 kJ mol-1. The effect
of Pt relaxation was greatest on the atop sites with the single Pt atoms
co-ordinated to the ethene moving 0.356 Å out of the surface for both
adsorption modes. This was accompanied by an increase of the adsorption energy
of 26 kJ mol-1 with the atop bridge (85.8 kJ mol-1)
slightly more stable than the atop hollow (84.8 kJ mol-1). The
hollow sites were effected by surface relaxation much so that the energetic
order of the atop and hollow sites is reversed when surface relaxation is
included indicating that the latter are unlikely to be observed.



It is clear from this study that if accurate energetics for surface adsorptions
and reactions are to be obtained then full surface relaxation must be included
in the calculations.
related references:
- Watson G.W. Wells R.P.K., Willock D.J and Hutchings G.J.
Density functional theory calculations on the interaction of ethene with
the {111} surface of platinum.
Journal of Physical Chemistry, 104, 6439-6446 (2000).
Email: watsong AT tcd.ie
Last updated: Apr 14 2012
Back to Top
|